Bacsnp是用来分析杆状病毒毒株之间遗传多样性(判断SNP的种群特异性)的下游工具。
安装
新建R环境
环境需求R 3.6.3, 严格限制在此版本,其他版本均存在依赖限制的问题。
因此,参照anaconda官方推荐,新建一个R 3.6.3的环境。
目前,官方conda源中的R似乎还没有更新到4.0.2, 最近的版本正是3.6.3. conda-forge或R的源中已更新至4.0.2
1
2
3
4
5
|
conda create -n R-3.6.3 r-essentials r-base
conda activate R-3.6.3
##如果你不小心将事实为R-3.6.3的环境命名成了R-4.0.2, 那么如下命令可以帮你将环境改回合适的名称
##conda create --name R-3.6.3 --copy --clone R-4.0.2
##conda remove --name R-4.0.2 --all # or its alias: `conda env remove --name R-4.0.2`
|
配置R软件源(可选)
目前官方源的范围速度还不错,因此也可直接使用官方源。
新建~/.Rprofile, 在其中添加如下语句。
1
2
3
4
|
options(repos=structure(c(CRAN=“https://mirrors.tuna.tsinghua.edu.cn/CRAN/”)))
source(“http://bioconductor.org/biocLite.R”)
options(BioC_mirror=“http://mirrors.ustc.edu.cn/bioc/”)
|
安装依赖
1
2
3
4
5
6
7
8
9
|
install.packages("devtools")
install.packages("ggplot2")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("VariantAnnotation")
## 确认这3个包成功安装后
library(devtools)
|
但在编译vignettes过程中报错:
1
2
3
4
5
6
|
Error: Failed to install 'unknown package' from GitHub:
System command 'Rcmd.exe' failed, exit status: 1, stdout + stderr (last 10 lines):
E> Quitting from lines 107-108 (bacsnp.Rmd)
E> Error: processing vignette 'bacsnp.Rmd' failed with diagnostics:
E> more cluster centers than distinct data points.
E> --- failed re-building 'bacsnp.Rmd'
|
需要修改源文件,在bacsnp.Rmd中的数据导入之前加入一行
1
|
options(stringsAsFactors = FALSE)
|
防止读入数据转化为data.frame时将字符串自动转化为因子。
然后,下载源码后手动本地安装
1
|
install_local("bacsnp-master.zip", build_vignettes = TRUE)
|
其实,不编译vignettes也是解决方案,install_github("wennj/bacsnp", build_vignettes = FALSE)即可。
数据预处理
bacsnp推荐的数据预处理模式如下。
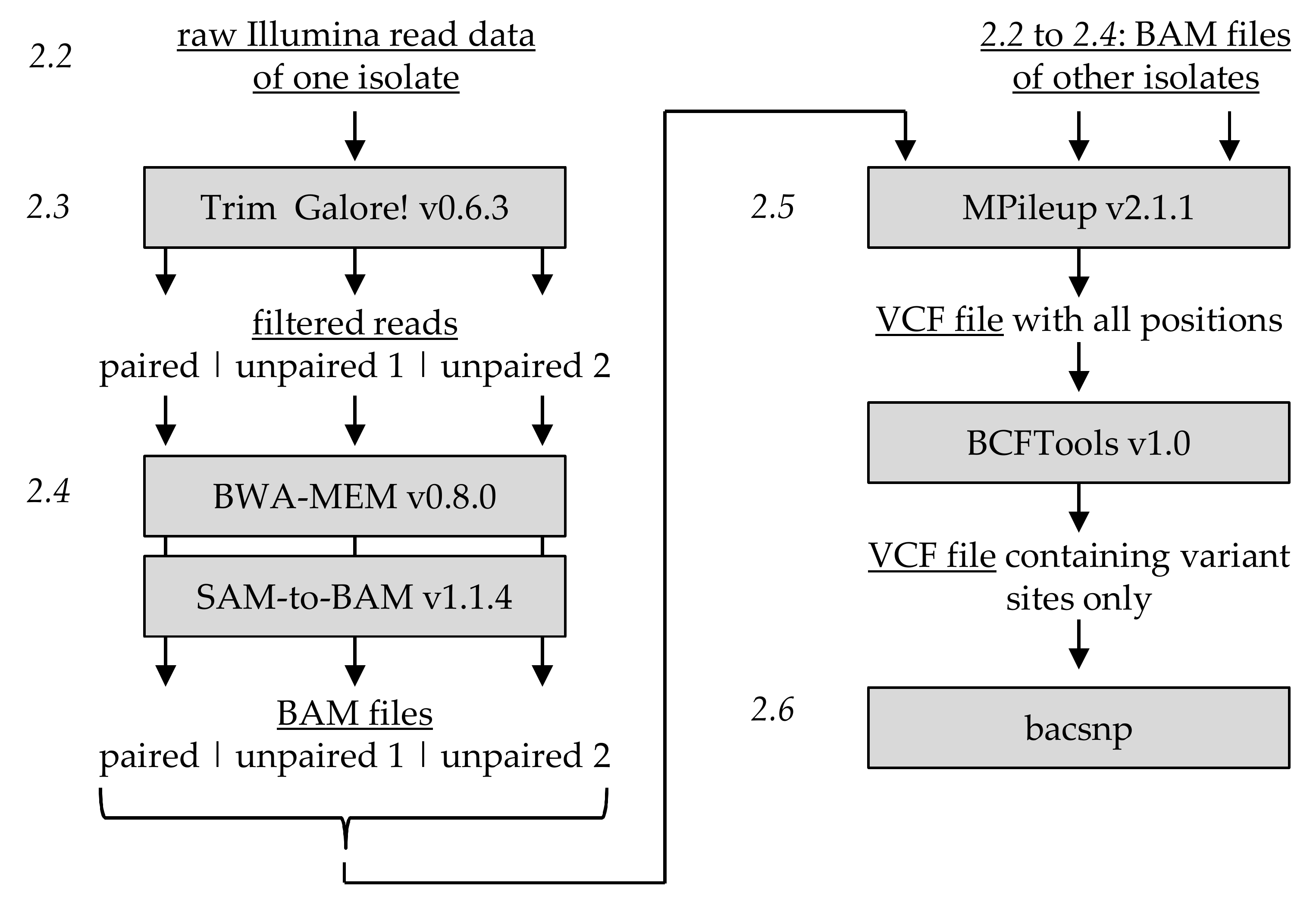
根据其vignettes中的bacsnp.Rmd文件中的详细说明和提供的bac.vcf示例,其详细要求如下:
- 只接受Illunima数据。(长读长的测序数据通过合适的比对工具也能够用于后续分析 )
- 测序数据应使用BWA比对至参考基因组,每个数据组要具有相应的标记。(经测试,bowtie2等其他工具也能够完成)
- 每个样品的数据都应当比对至同一个参考基因组序列
- 使用samtool/bcftool mpileup来分析BAM文件(不进行indel分析)
- bcftools只用来获取变异位点
- bcftool的输出文件为非压缩的VCF格式
观察示例的bac.vcf文件,发现其Galaxy服务端的环境配置如下。
1
2
3
4
5
6
|
##samtoolsVersion=1.8+htslib-1.8
##samtoolsCommand=samtools mpileup -f /CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540916.dat -C 0 -d 328 -q 0 -Q 13 --VCF --uncompressed --output-tags DP,DPR -I --output /CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540952.dat /CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540935.dat /CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540936.dat /CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540937.dat /CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540949.dat /CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540950.dat /CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540951.dat
##reference=file:///CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540916.dat
......
##bcftools_callVersion=1.2+htslib-1.2.1
##bcftools_callCommand=call -O v -A -v -f GQ -m -o /CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540976.dat /CLUSTER-FS/galaxy-dist/database/files/000/540/dataset_540952.dat
|
即使用samtool 1.8与bcftool 1.2的组合。作者强调samtool mpileup的输出文件需要有DP,DPR两个tag。但是,samtool在1.9之后的版本用AD替换了DPR,所以除非手动修改结果VCF文件,那么还是按照作者的环境配置来进行更省事。
bacsnp分析
将bac.rmd的内容摘录如下
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
|
library(VariantAnnotation)
library(IRanges)
library(bacsnp)
## Loading VCF file data
bacdata <- readVcf("bac.vcf")
## Transforming VCF file data
bac.df <- bacsnp.transformation(bacdata)
head(bac.df)
## Accessing basic SNP information
unique(bac.df$ISO)
length(unique(bac.df$POS))
unique(bac.df$POS)
## Filtering SNP data frame
bac.f <- bacsnp.filter(bac.df, min.abs.cov = 100, min.abs.alt = 10, min.rel.alt = 0.05)
## Determination of SNP specificity
bac.spec <- bacsnp.specificity(bac.f, c("iIso1", "iIso2"), which.rel = "REL.ALT1")
bac.spec$combinations
head(bac.spec$data[, c("POS", "REF", "ALT","REL.ALT1", "SPEC", "GROUP.ID", "NT.SPEC")])
## Visualization of SNP frequencies with bacsnp.plot
i1 <- bac.spec$data[bac.spec$data$ISO == "iIso1",]
i2 <- bac.spec$data[bac.spec$data$ISO == "iIso2",]
bacsnp.plot(i1, col = "SPEC", genome.length = 123193, mark.repeats = FALSE)#, mark.lowGQ = 120, mark.lowQUAL = 400)
bacsnp.plot(i2, col = "SPEC", genome.length = 123193, mark.repeats = FALSE)#, mark.lowGQ = 120, mark.lowQUAL = 400)
|
结果
这个工具以多个毒株(种群)的二代重测序数据作为输入,能够分析得到种群特有的(或多个种群共有的)SNP位点,并进行可视化。
可视化过程实际是从第一个SNP位点(基于基因组位置)开始。如果样本总体的SNP位点不多,在基因组起始5 kb的位置内又没有SNP位点,那么结果图中就不是从0 bp开始展示整个基因组的情况。作者在bacsnp.plot()中加入了genome.length参数输入基因组总长度,保证能够绘制到基因组的末尾,却忘了不一定会有开头。
参考来源
Wennmann, Jörg T.; Fan, Jiangbin; Jehle, Johannes A. 2020. “Bacsnp: Using Single Nucleotide Polymorphism (SNP) Specificities and Frequencies to Identify Genotype Composition in Baculoviruses." Viruses 12, no. 6: 625.
http://samtools.github.io/bcftools/howtos/variant-calling.html
https://github.com/wennj/bacsnp/blob/master/vignettes/bacsnp.Rmd
